- Asociación Síndrome 22q11 / Entidad de Utilidad Pública


Por síndrome de deleción 22q11 se entiende un patrón de anomalías que se producen cuando se pierde un fragmento o región específica del cromosoma 22, denominada precisamente 22q11.2.
La región cromosómica 22q11.2 alberga medio centenar de genes, algunos de los cuales controlan determinados aspectos del desarrollo embrionario.
Específicamente, la migración de células originarias de una zona del esbozo del sistema nervioso central conocida como “cresta neural”, que intervienen en el desarrollo de una serie de estructuras como el timo, la glándula paratiroides y el corazón (ver figura 1). Estas células también participan en el desarrollo del paladar y de determinadas zonas del cerebro.
El timo es un órgano cuya misión fundamental es programar a una población de glóbulos blancos que produce la médula ósea (linfocitos T), para que reconozcan diferentes proteínas del organismo como propias o extrañas y dirijan la respuesta inmunitaria a infecciones.
La glándula paratiroides regula los niveles sanguíneos de calcio, movilizando las reservas del organismo cuando los niveles bajan, o disminuyendo su absorción intestinal y promoviendo su eliminación renal cuando los niveles suben.
La formación del corazón es muy compleja. Se inicia a partir de una estructura en forma de tubo que se pliega sobre sí misma y posteriormente se tabica para dar lugar a las distintas cámaras del corazón (ventrículos y aurículas) y a los grandes vasos que salen de estas (troncos aórtico y pulmonar, y venas cavas).
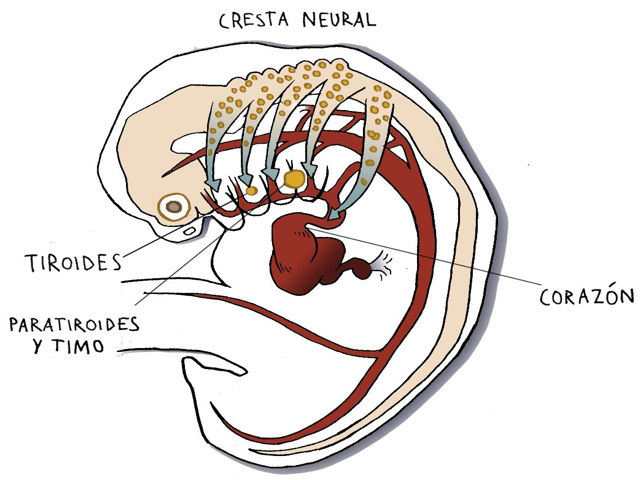
Figura 1. Migración de las células de la cresta neural en un embrión de 4-6 semanas.
Ilustración de Lucas García-Miñaúr, basada en Scambler PJ. The 22q11 deletion síndromes. Human Molecular Genetics 2000; 9:2421-26
La deleción 22q11 es la anomalía cromosómica submicroscópica más frecuente, con una frecuencia estimada en la población general de 1 en 4000 recién nacidos, aunque muy posiblemente la frecuencia real sea mayor.
El motivo de esta alta frecuencia y de que el tamaño del fragmento de cromosoma que se pierde sea siempre el mismo en la mayoría de los casos (95%) se debe a la estructura del ADN de esta región, flanqueada por elementos repetitivos de ADN prácticamente idénticos, que predispone a que se produzca este tipo de accidente (reestructuraciones, tanto por defecto -deleciones- como por exceso -duplicaciones-) en la formación de las células reproductivas (espermatozoides y óvulos).
Lo que no se ha logrado explicar aún es la gran variabilidad de las manifestaciones clínicas entre personas que han perdido la misma cantidad de material genético. Se considera que existen otros factores genéticos, no identificados por el momento, que modifican el impacto de la deleción en cada persona. Es de esperar que estos factores se vayan conociendo progresivamente en los próximos años.
¿Qué manifestaciones clínicas y problemas médicos se asocian con el síndrome de deleción 22q11?
Las manifestaciones del síndrome de deleción 22q11 se relacionan principalmente con las diferentes estructuras y órganos afectados:
La presencia o no de estos problemas y su gravedad varía de unas personas a otras. Y tampoco se relacionan entre sí; es decir, la existencia de una anomalía cardiaca no implica una mayor probabilidad de padecer problemas del paladar o retraso del desarrollo psicomotor.
Además de estos problemas médicos, que tienen una mayor relevancia en los primeros años de vida, las personas con síndrome de deleción 22q11 presentan particularidades en su desarrollo psicomotor y dificultades de aprendizaje muy específicas, que se van conociendo y definiendo mejor en los últimos años.
Es muy habitual el retraso en el desarrollo del lenguaje (80%), independientemente de que exista o no un problema asociado del paladar.
En el periodo escolar se suele manifestar con frecuencia un déficit de la memoria de trabajo que dificulta el procesamiento de la información y el razonamiento abstracto necesario para resolver problemas, comprender bien lo que se lee y el cálculo matemático. A pesar de que estos déficits son muy específicos, pueden pasar desapercibidos a los propios profesores.
Son también frecuentes los trastornos del comportamiento, como el déficit de atención, la ansiedad, el trastorno de oposición desafiante y el obsesivo-compulsivo. Todo ello hace a estas personas más vulnerables en el entorno escolar, sobre todo en momentos de mayor exigencia académica y social como es la adolescencia.
Estos problemas de aprendizaje e integración social causan una gran inquietud a sus familias. La deleción 22q11 predispone también a la aparición de trastornos psiquiátricos en la edad adulta. La frecuencia de esquizofrenia en adultos con síndrome de deleción 22q11 es 20 veces superior a la de la población general.
Ninguna.
Todos se refieren al mismo trastorno genético.
Lo que ocurre es que se describieron de forma independiente como síndrome de DiGeorge, síndrome de Shprintzen o velo-cardio-facial, y síndrome de la cara de la anomalía troncoconal, pensando que se trataban de trastornos diferentes.
Con el tiempo se ha comprobado que la causa de todos ellos es la misma: la pérdida o deleción de la región cromosómica 22q11.2.
El motivo de esta confusión de nombres se debe a que cada uno de los médicos que los describieron hacía más hincapié en determinadas manifestaciones clínicas, de acuerdo con su especialidad. Así, Angelo DiGeorge era endocrinólogo infantil, Robert Shprintzen cirujano especialista en problemas del paladar, y Atsuyoshi Takao cardiólogo infantil.
Buscando un término común, John Burn, genetista clínico británico, propuso el acrónimo CATCH-22 (Cardiac Abnormality Thymus Calcium Heart-22); pero en el mundo anglosajón la expresión “situación CATCH-22”, basada en la célebre novela de Joseph Heller, alude a una situación sin salida, o de pescadilla que se muerde la cola, por lo que no fue bien recibida y terminó por desecharse.
La tendencia actual es a utilizar el término síndrome de deleción 22q11. Existe otra deleción en una región más distal del cromosoma 22, conocida como deleción 22q13, menos frecuente y con otro tipo de manifestaciones clínicas con la que no se debe confundir.
A pesar de que no se puede reponer esta pérdida de material genético y no existe una cura como tal, un seguimiento médico y educativo dirigido puede ayudar a prevenir, mejorar y corregir muchos de los problemas asociados y conseguir que estos niños se sientan más integrados, rindan más y sean más felices.
Dado que el síndrome de deleción 22q11 afecta a diferentes órganos y sistemas, es recomendable que la atención de estas personas sea llevada a cabo por un equipo multidisciplinar que incluya un núcleo básico compuesto por especialistas en cardiología, inmunología, foniatría, otorrinolaringología (ORL), cirugía maxilofacial (CMF), endocrinología y genética clínica.
Dependiendo de cada caso puede ser necesario consultar con otros especialistas (neurología, ortopedia, nefrología, oftalmología, etc.). Es muy valioso contar con especialistas en psicología clínica y psiquiatría que puedan hacer una valoración neurocognitiva de estas personas, idealmente al final de la primera infancia, a partir de los cuatro años de edad, y organizar el seguimiento.
También es deseable implicar al equipo de orientación escolar, de modo que conozcan el diagnóstico y las necesidades educativas especiales de estos niños. La atención médica resultará más eficiente si es coordinada por un pediatra o por un genetista clínico familiarizado con el síndrome de deleción 22q11.
Aspectos médicos: Recomendaciones de valoración inicial y seguimiento
Se han publicado recientemente recomendaciones y guías de siguimiento médico para personas con Síndrome de deleción 22q11. Algunas de éstas las podéis encontrar en en nuestro apartado de Profesionales (pinchar aquí)
Recomendaciones fundamentales.
La incorporación de nuevas técnicas moleculares, como el MLPA o la hibridación genómica comparada con arrays (array-CGH) ha permitido la detección progresiva de duplicaciones de la región 22q11.2. La duplicación es la anomalía cromosómica complementaria a la deleción, y se produce al mismo tiempo que ésta.
El problema es que sus manifestaciones son habitualmente más leves y menos reconocibles, por lo que puede pasar totalmente desapercibida. La duplicación es muy difícil de detectar mediante la técnica FISH, que ha supuesto durante muchos años la forma habitual de diagnosticar la deleción 22q11.2.
Las nuevas técnicas moleculares (MLPA, array-CGH), basadas en la medición de la cantidad de ADN, sin necesidad de depender de la visualización de una señal luminosa en una región cromosómica determinada (FISH), por otro lado muy difícil de cuantificar, detectan con gran precisión tanto la pérdida (deleción) como la ganancia (duplicación) de material cromosómico de la región 22q11.2.
Al contrario de lo que sucede con la deleción, en la mayoría de cuyos casos se puede sospechar este diagnóstico por el patrón de problemas médicos y ciertos rasgos faciales característicos, las duplicaciones se identifican en pacientes estudiados por diferentes motivos, que también se asocian a la deleción, pero que de forma aislada son poco específicos (talla baja, dificultades de aprendizaje, anomalías cardiacas congénitas, etc.).
Como sucede con la deleción, las manifestaciones clínicas son muy variables, y en una gran proporción de casos estas duplicaciones se han heredado de un progenitor sin manifestaciones aparentes o tan leves que han pasado desapercibidas.
Todo esto es bastante reciente, por lo que aún se desconoce con exactitud el abanico de problemas médicos que puede producir la duplicación y sus efectos a largo plazo. Los profesionales implicados en la valoración y seguimiento de estos pacientes son los mismos que atienden a pacientes con la deleción.
Por todos estos motivos, consideramos es necesario incluir a esta anomalía cromosómica “hermana” dentro de la asociación 22q.
Tu ayuda es clave para dar a conocer el síndrome y mejorar la calidad de vida de niños y adultos afectados.
Las colaboraciones con nuestra asociación son fundamentales para poder seguir difundiendo el síndrome 22q.
Si deseas colaborar puedes hacerlo también adquiriendo alguno de nuestros regalos solidarios de 22q.
Si eres una persona con espíritu solidario y te apetece aprender de las personas con una realidad diferente a la tuya.